Protein PhylogenyWhat is phylogeny?Phylogeny is a study of evolution that focuses on the relatedness of species based on a common ancestor. To understand relatedness between species, amino acid sequence comparisons can provide insight into the molecular alterations that have accumulated within species and are used to identify changes that occurred after the divergence of the species. To visualize the cumulative variation among organisms, phylogentic trees can be constructed, illustrating the relatedness among species or individual proteins and the divergence of species throughout time.
Sequence AlignmentTo begin the process of creating phylogenetic trees, the sequences of the proteins of each organism to be studied must be aligned and scored to determine the degree to which the sequences are identical. Clustal Omega is an online multiple sequence alignment program that provides a range of outputs to analyze sequence alignments and was used to align and score the FANCL homologs [1].
Clustal Omega requires FASTA formatted sequences to be entered for each protein of interest. These FASTA sequences for the FANCL homologs were obtained from the NCBI gene database and are compiled in the document below. Upon completion of the alignment, the program provides a complete alignment of the protein sequence that can be modified to highlight multiple aspects of the similarities and differences at each amino acid position in the sequence. Below is the sequence alignment for the human FANCL protein and the nine homologous proteins. 
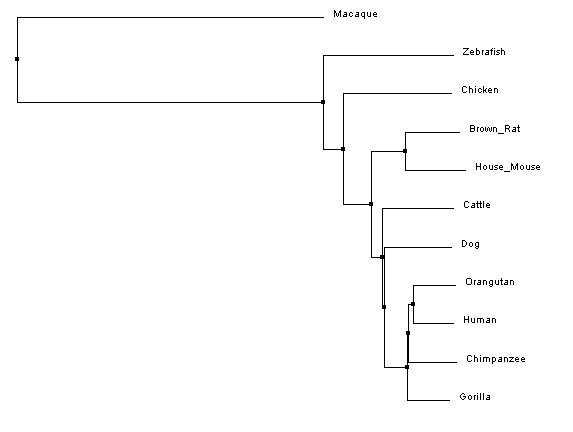
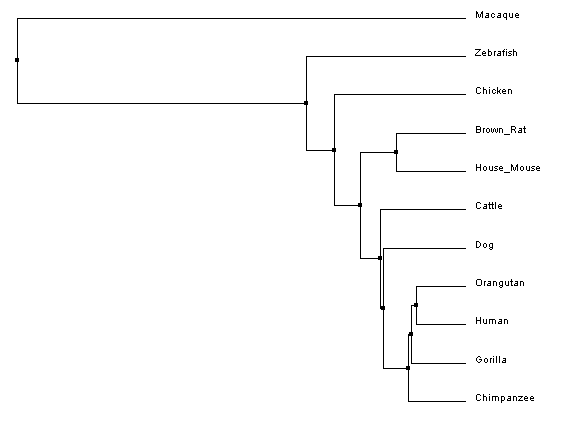
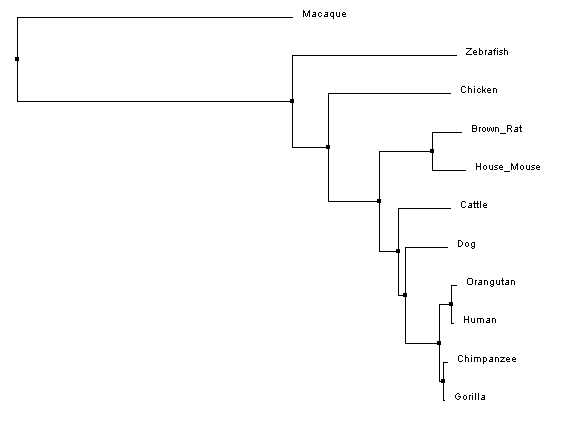
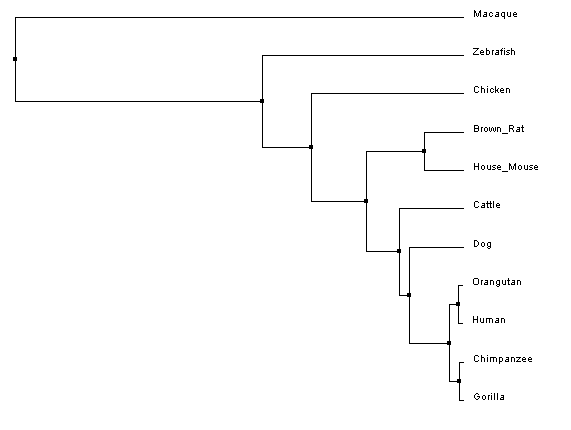
Constructing Phylogenetic TreesAfter aligning sequences, phylogenetic trees can be constructed to predict the relatedness of each protein to the others. Clustal Omega calculates these phylogenetic trees directly within the program used to align the sequences, and can predict trees using two different similarity scoring methods in combination with two different tree construction models.
Analysis of ResultsUnlike the nucleotide sequence phylogenetic analysis, the protein analysis provides a much more predictable outcome, with protein similarity branching matching predicted species divergence patterns; however, like the nucleotide trees, the rhesus macaque is an outlier. After looking at the protein sequence alignment and back to the nucleotide sequence alignment, it was discovered that a single mutation cause a premature start codon nearly ninety amino acids upstream of the typical start codon for the FANCL protein. Although the the remainder of the sequence is in line with, and has higher sequence identity to, the primate homologs, the ninety amino acid addition to the beginning of the protein greatly affects the overal sequence identity, resulting in the aberrant placement of the macaque in the phylogenetic tree.
|
References
[1] Sievers F, Wilm A, Dineen DG, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DMolecular Systems Biology 7 Article number: 539 doi:10.1038/msb.2011.75
[2] Barton, N. H., Briggs, D. E., Eisen, J. A., Goldstein, D. B., & Patel, N. H. (2007). Evolution. N.p.: Cold Spring Harbor.
[2] Barton, N. H., Briggs, D. E., Eisen, J. A., Goldstein, D. B., & Patel, N. H. (2007). Evolution. N.p.: Cold Spring Harbor.
File Resources
| fancl.txt |
